Mastocitosi cutanea
SALERNO
- MASTOCITOSI CUTANEA E SISTEMICA
- Le mastocitosi sono un gruppo di patologie rare caratterizzate dall’accumulo di mastociti nei tessuti. In alcune mastocitosi cutanee, i mastociti si accumulano in numero eccessivo nel derma, causando rossore, pomfi e prurito, per la liberazione di svariati mediatori chimici (es. istamina, prostaglandine PGD2, leucotrieni LC4, fattore attivante le piastrine PAF, triptasi, chimasi, VEGF, eparina, fosfolipasi A2, citochine, etc). Nelle mastocitosi sistemiche, i mastociti possono accumularsi in uno o più organi (es. ossa, apparato gastrointestinale, midollo osseo, etc), con sintomi più o meno gravi in base al tipo di organo interessato.
LE CAUSE DELLA MASTOCITOSI
Tra le cause di mastocitosi sono state riportate diverse mutazioni somatiche puntiformi del proto oncogene c-kit che codifica per il recettore di membrana dei mastociti per il fattore di crescita stem cell factor (una delle mutazioni più frequenti è detta D816V e si verifica a livello del codone 816 che codifica per questa importante tirosin chinasi), che indurrebbe una proliferazione clonale (indipendente dallo stem cell factor SCF) con iperplasia e accumulo tissutale di queste cellule. I mastociti sono cellule del sistema immunitario tissutale e partecipano attivamente nell’infiammazione, potendo essere attivati da svariati fattori in grado di interagire con specifici recettori presenti sulla superficie dei mastociti (es. recettore c-kit, recettore per le IgE, recettori vanilloidi, etc). La cute umana contiene circa 7000 mastociti per mm quadrato, distribuiti prevalentemente intorno ai vasi dermici (nei processi infiammatori, i mastociti favoriscono il transito dei granulociti ematici attraverso gli endoteli, grazie alla produzione di sostanze vasoattive come eparina e istamina), intorno agli annessi cutanei e intorno alle fibre nervose dermo epidermiche. Infatti i mastociti sono spesso coinvolti nella genesi del prurito neuropatico associato a nevralgia delle piccole fibre terminali del sistema nocicettivo cutaneo (neuropatie dermoepidermiche). I mastociti di cute (chimasi positivi) e mucose (chimasi negativi) svolgono un ruolo strategico come coordinatori della soglia del dolore neuropatico, possendendo recettori vanilloidi di membrana TRPV1, analogamente ai neuroni sensitivi e sono coivolti in molte allodinie dermoepidermiche, tra cui il lichen simplex, la vulvodinia, l’acquadinia e sindrome della bocca urente. Nei processi infiammatori cronici, la sostanza P (sostanza responsabile delle sensazioni dolorose) si lega al recettore vanilloide TRPV1 sulla superficie dei mastociti, inducendone la degranulazione, con liberazione di istamina e di altra sostanza P, che amplifica il processo flogistico. In queste situazioni i mastociti producono fattori di crescita per i vasi sanguigni (VEGF) e per le terminazioni nervose (NGF), con azione proinfiammatoria sull’angiogenesi e sulla produzione di neuropeptidi, a cui può far seguito un abbassamento della soglia del dolore.
L’ORTICARIA PIGMENTOSA È UNA MASTOCITOSI CUTANEA
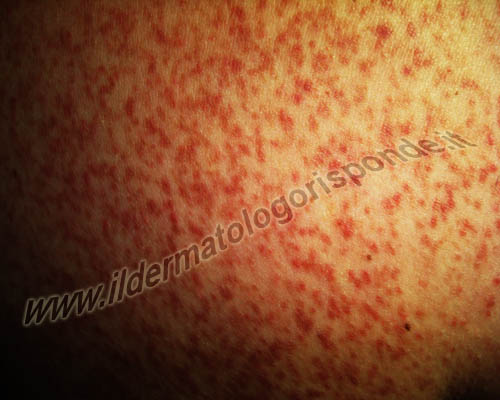
La mastocitosi cutanea più frequente è l’orticaria pigmentosa, caratterizzata dalla comparsa di macule o papule brunastre, al tronco, agli arti e meno frequentemente al viso. Essa può esordire nei bambini, anche molto piccoli, così come negli adulti, con pomfi, prurito e persino bolle, nelle aree sottoposte a sfregamento (segno di Darier). Si chiama orticaria pigmentosa per l’accumulo di melanina nello strato basale dell’epidermide, ma in presenza di macule pigmentate è importante ricercare ed escludere mediante un checkup mirato, eventuali condizioni che possono entrare in diagnosi differenziale con questo tipo di mastocitosi (es. chiazze caffelatte, neurofibromatosi, etc). La mastocitosi teleangectasica (teleangectasia macularis eruptiva perstans di Weber e Hellenschmied) è invece più frequente negli adulti e si presenta con macule rossastre in cui manca il segno di Darier (il segno di Darier della mastocitosi non va confuso con la discheratosi follicolare della malattia di Darier). Alcune forme di mastocitosi cutanee (es. mastocitosi bollosa, mastocitosi a chiazze, mastocitosi nodulare, mastocitoma solitario di Tilbury Fox, etc) sono più frequenti in età pediatrica. Il mastocitoma solitario, fu descritto già nel XIX secolo dal dermatologo inglese William Tilbury Fox, ma come entità autonoma fu descritto in dettaglio nel 1954 dal patologo scozzese Alexander James Murray Drennan e dal dermatologo inglese John Martin Beare. Esso si presenta nel bambino, alla nascita o nei mesi successivi, come nodulo di colorito variabile tra il rosso e il marrone e autorisolve nella maggior parte dei casi nel giro di alcuni anni. Va differenziato da altre neoformazioni cutanee dell’età pediatrica (per esempio nevo di Spitz, mollusco contagioso, granuloma piogenico) e talora presenta una positività al cosiddetto segno di Darier. Le mastocitosi cutanee diffuse, si presentano invece con una cute diffusamente ispessita e di consistenza pastosa. In questi casi, l’imbrunimento della cute è dovuto a un’iperplasia reattiva dei melanociti, oltre che dei mastociti, mediata dai recettori transmembrana c-kit situati sulla superficie di queste cellule. La degranulazione dei mastociti si può manifestare con prurito, vampate di calore e altri sintomi potenzialmente recidivanti (per esempio nausea, dispnea, cefalea, diarrea, palpitazioni, ipotensione, sincope, anafilassi).

La mastocitosi cutanea può presentarsi come orticaria pigmentosa o come mastocitosi maculare di Weber e Hellenschmied
LE MASTOCITOSI SISTEMICHE POSSONO INTERESSARE UNO O PIÙ ORGANI
Le mastocitosi sistemiche sono meno frequenti delle forme cutanee e vanno ricercate mediante un checkup mirato, che comprende tra i vari esami una mineralometria ossea (MOC), una TAC, una radiografia completa dello scheletro o una scintigrafia ossea (mastocitosi ossea), una esofagogastroduodenoscopia e un’ecografia epatosplenica (mastocitosi dell’apparato digerente), un prelievo ematico (per esempio emocromo completo con formula, sideremia, ferritina, AST, ALT, ANA, fosfatasi alcalina, colesterolo, trigliceridi, istaminemia, complementemia, dosaggio IgE totali, dosaggio della triptasi, etc), un esame delle urine (es. dosaggio della N-metilistamina, dell’acido N-metilimidazolacetico e dei metaboliti urinari delle prostaglandine) e una biopsia dell’organo sospetto (es. midollo osseo, fegato, etc). Tra i vari esami, rivestono particolare importanza la biopsia osteomidollare (ricerca della mutazione del gene c-kit, ricerca di infiltrati mastocitari multifocali, osteosclerosi, fibrosi midollare, citofluorimetria, etc) e il dosaggio della triptasi sierica (enzima prodotto dai mastociti che circola nel sangue di tutti gli individui in concentrazioni inferiori ai 15 ng/ml e che può aumentare in alcune forme di mastocitosi). La biopsia osteomidollare è importante nei pazienti con livelli molto elevati di triptasi. In questi pazienti, la triptasemia può rappresentare un parametro utile nel followup, come indice indiretto di risposta alla terapia. Nella maggior parte delle mastocitosi cutanee, i livelli ematici di triptasi sono normali, i mastociti presentano caratterisiche morfologiche apparentemente normali, mentre in alcune forme sistemiche, essi possono avere una morfologia allungata ed esprimere in superficie dei marcatori normalmente non presenti sui mastociti normali (es. CD2, CD25, etc). Le forme sistemiche richiedono nella maggior parte dei casi un approccio diagnostico terapeutico di tipo multidisciplinare (es. ematologo, immunologo, internista, oncologo, etc).
DECORSO CLINICO E TERAPIA DELLE MASTOCITOSI
Le mastocitosi cutanee del bambino hanno solitamente un decorso favorevole con possibile regressione dei sintomi durante l’adolescenza, ma un checkup è utile per riconoscere eventuali forme sistemiche, specie in caso di persistenza nell’età adulta. Nell’adulto la remissione spontanea è meno frequente. Quando i mastociti, pur essendo in numero normale, diventano particolarmente sensibili agli stimoli ordinari, si parla di sindrome da attivazione mastocitaria o MCAS (Mast cell activation syndrome). Per quanto riguarda la terapia della mastocitosi, essa comprende a seconda dei casi, l’impiego di eventuali farmaci (per esempio antistaminici, steroidi sistemici, antileucotrienici, inibitori delle lipossigenasi, sodio cromoglicato, interferone alfa, 2-cloro-deossiadenosina, cladribina, imatinib, etc) sotto la guida del proprio medico, evitando le possibili cause di degranulazione mastocitaria (per esempio farmaci come acido acetilsalicilico, cibi istamino liberatori come formaggi, pesci e crostacei, punture di imenotteri, etc). Alcuni farmaci inibitori delle tirosin chinasi, risultano meno efficaci nei pazienti portatori della mutazione D816V.
LE MASTOCITOSI NELLA STORIA DELLA MEDICINA
L’orticaria pigmentosa viene descritta nel 1869 dal dermatologo inglese Edward Nettleship e dall’oftalmologo inglese Warren Tay come una rara forma di orticaria. Nel 1875 il dermatologo inglese William Tilbury Fox descrive la mastocitosi xantelasmoidea, nota anche come mastocitosi pseudoxantomatosa a placche. Nel 1876 il dermatologo americano Prince Albert Morrow, descrive l’orticaria pigmentosa come eritema tubercolare. Il 17 giugno 1878, il medico tedesco Paul Ehrlich descrive per la prima volta il mastocita durante la sua tesi di laurea, ricevendo il premio Nobel per la medicina nel 1908. Sempre nel 1878, il dermatologo inglese Alfred Sangster del Charing Cross Hospital di Londra, utilizza per la prima volta il termine di urticaria pigmentosa per descrivere la malattia descritta 9 anni prima da Nettleship e Tay. Nel 1883 il dermatologo inglese William Tilbury Fox introduce in letteratura il termine orticaria xantelasmoidea. Nel 1887 il dermatologo tedesco Paul Gerson Unna scopre che l’orticaria pigmentosa è dovuta a un accumulo di mastociti nel derma. Il termine mastocitosi venne utilizzato nel 1936 dal dermatologo francese Albert Sézary per indicare le forme cutanee diffuse, mentre la prima forma di mastocitosi sistemica non cutanea, è stata riportata nel 1949 dal Dr. John Ellis. Nel 1950 viene riportato il primo caso di leucemia mastocitaria. Alla fine degli anni ’70, viene riportato il miglioramento dei pazienti affetti da orticaria pigmentosa trattati con PUVA terapia. Nel 2001, l’Organizzazione Mondiale della Sanità, pubblica una classificazione molto dettagliata delle varianti cliniche della malattia, suddividendole in mastocitosi cutanee (es. orticaria pigmentosa, orticaria diffusa, mastocitoma solitario di Tilbury Fox, etc), mastocitosi sistemiche indolenti (es. mastocitosi del midollo osseo), mastocitosi sistemiche subacute, mastocitosi sistemiche associate ad anomalie ematologiche clonali non mastocitarie, mastocitosi sistemiche aggressive e altre patologie mastocitarie (es. mastocitoma extracutaneo, sarcoma mastocitario, leucemia mastocitaria). Nell’attuale classificazione internazionale delle malattie ICD-11 il mastocitoma cutaneo è riportato con il codice XH1J17, l’orticaria pigmentosa con il codice XH74F2, e la mastocitosi cutanea diffusa con il codice XH2RG8.
